
EUの医療機器業界は、医療機器指令(MDD)から医療機器規則(MDR)への移行に伴い、大きな変革を遂げました。この変化は、EU市場における医療機器の安全性、性能、追跡性を向上させることを目的としています。規制の専門用語に馴染みのない方々に向けて、MDDとMDRの主な違いをわかりやすく説明します。
MDDとは?
医療機器指令(MDD)は、1993年に導入され、EU加盟国間で医療機器に関する規制要件を調和させることを目的としていました。その主な目的は、EUで販売される医療機器が高い安全性と性能を満たすことを保証することでした。MDDのもとでCEマークを取得した製品は、欧州経済領域(EEA)全域で販売可能でした。
MDRとは?
医療機器規則(MDR)は、2017年5月26日に施行され、2021年5月26日に移行期間が終了しました。MDRは、MDDの欠点を解消し、技術の進歩や安全性の向上に対応するために導入されました。EU市場における医療機器の安全性、性能、品質を向上させるためのより強固な規制枠組みを提供することを目指しています。
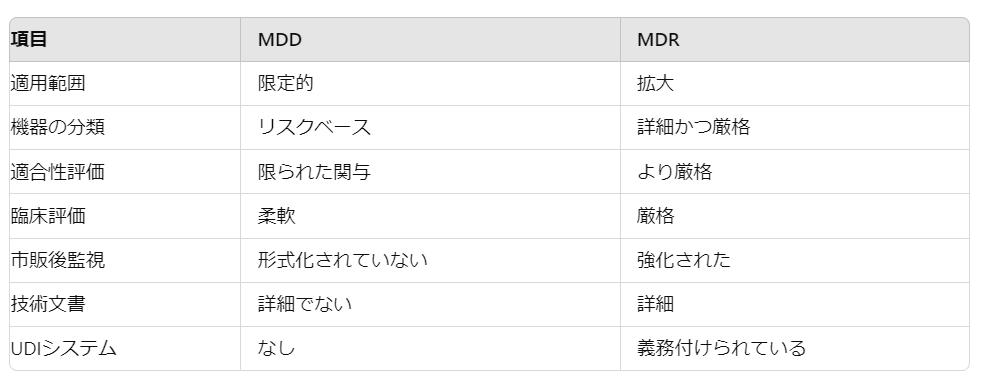
MDDとMDRの主な違い
- 適用範囲と定義:
- MDD: 適用範囲は狭く、主に医療機器と能動埋め込み型医療機器を対象としていました。一部の製品(例:化粧品デバイスや独立型ソフトウェア)に対する規定は限定的でした。
- MDR: 適用範囲が大幅に拡大され、清掃、消毒、滅菌用の機器や、化粧品デバイス、カラーコンタクトレンズなど医療目的でないデバイスも明確に対象としています。また、医療目的で使用される独立型ソフトウェアも含まれます。
- 機器の分類:
- MDD: リスクに基づいて機器を分類し、クラスI(低リスク)からクラスIII(高リスク)までのカテゴリーがありましたが、分類ルールは曖昧であることがありました。
- MDR: より詳細かつ厳格な分類ルールが導入されました。特定の機器は、より高いリスクカテゴリーに再分類され、より厳格な適合性評価が求められます。例えば、再使用可能な手術器具はクラスIからクラスIIaに再分類されました。
- 適合性評価:
- MDD: 適合性評価手続きは機器のクラスによって異なり、Notified Bodies(認証機関)が重要な役割を果たしていましたが、低リスク機器ではその関与が限定的でした。
- MDR: 特に高リスク機器に対してより厳格な適合性評価要件が課され、Notified Bodiesの関与が増加します。また、ほとんどの機器に対して臨床評価と市販後臨床フォローアップ(PMCF)の要件が導入されました。
- 臨床評価と調査:
- MDD: 臨床評価は要求されていましたが、既存の文献や同等機器に依存する柔軟性がありました。臨床調査の定義は曖昧でした。
- MDR: より厳格な臨床評価要件が設定され、機器に特有の臨床データの必要性が強調されています。特に高リスク機器や新技術に対しては包括的な臨床調査が必要です。
- 市販後監視と警戒:
- MDD: 市販後監視(PMS)と警戒システムは形式化されておらず、メーカー間での実施が一貫していませんでした。
- MDR: PMSおよび警戒要件が大幅に強化され、メーカーは積極的なPMSシステムを確立する必要があります。また、市販後パフォーマンスフォローアップ(PMPF)も導入されています。
- 技術文書:
- MDD: 技術文書の要件は詳細ではなく、提出物の品質や完全性にばらつきがありました。
- MDR: 技術文書に対して包括的で詳細な要件が設定され、設計、リスク管理、臨床評価、PMS活動に関する詳細な情報を含む技術文書ファイル(TDF)の維持が求められます。
- ユニークデバイス識別(UDI):
- MDD: UDIシステムの導入は義務付けられておらず、機器の追跡性や識別に課題がありました。
- MDR: UDIシステムが義務付けられ、追跡性と透明性が向上します。各機器には一意の識別子が付与され、供給チェーン全体での追跡が容易になり、効果的なリコールが可能になります。
- 経済事業者:
- MDD: 経済事業者に対する規定は限定的で、主にメーカーとその義務に焦点を当てていました。
- MDR: すべての経済事業者(メーカー、認可代表者、輸入業者、販売業者)に対する具体的な役割と責任が定義され、供給チェーン全体での機器の安全性とコンプライアンスを確保するための厳格な要件が課されます。
- EUDAMEDデータベース:
- MDD: EUDAMEDの役割と機能は限定的で、規制目的に完全には利用されていませんでした。
- MDR: EUDAMEDの役割と機能が拡大され、機器、経済事業者、臨床調査、警戒活動に関する情報を含む包括的なデータベースになります。これにより透明性が向上し、利害関係者間の情報交換が促進されます。
なぜ変わったのか?
MDDからMDRへの移行は、MDDの限界に対処し、医療技術の進歩に対応するために行われました。MDRは、より包括的で一貫した規制枠組みを提供し、EU市場における医療機器の安全性と性能を高めることを目指しています。
メーカーへの影響
メーカーは、新しくより厳格なMDRの要件を遵守するために、自社の製品を再評価し、技術文書を更新し、強固なPMSシステムを実施し、UDIシステムを通じて追跡性を確保する必要があります。この移行は多くのメーカーにとって挑戦的であり、要件を満たすために延長やサポートを求めるケースも見られます。
結論
MDDからMDRへの移行は、EUにおける医療機器の規制における重要な進化を示しています。MDRは、より厳格な要件を導入する一方で、最終的には医療機器の安全性、性能、追跡性を向上させ、患者や医療提供者に利益をもたらすことを目指しています。これらの変化を理解することで、医療機器業界の関係者は新しい規制環境に適切に対応し、EUにおける医療機器の安全性と性能の向上に貢献することができます。
これまでのMDDとMDRの主な違いと新要件について理解し、それに備えることで、規制の複雑さを乗り越え、コンプライアンスを確保することができます。
投稿者
ryuta@socraticincorporated.com
関連投稿





